domingo, 29 de julho de 2012
conclusão
Considero portanto que neste portfólio de biologia foram expostos vários temas fundamentais para se compreender melhor como age a genética nos dias atuais,a descoberta de várias tecnologias genéticas no melhoramento de organismos vivos,o avanço na recombinação do DNA que fizeram surgir técnicas a partir da engenharia genética,surgiram neste portfolio várias idéias sobre organismos geneticamente modificados,e sobre a sua manipulação na genética por meio de tecnológias de DNA recombinante,constou também neste portfolio algumas explicações de várias doenças humanas,tais como a endogamia,a fenilcetonúria,a galactosemia,o hipotireoidísmo congênito,a talassemia e a doença de tay-sachs,dentro dessas doenças também foram apresentados o diagnostico,como elas agem no organismo humano,e sobre o seu tratamento para melhor vida dos pacientes afetados.Para finalizar foram acrescentadas vários tipos de síndromes,tais como elas se desenvolvem,como elas atuam no organismo humano e se há cura para essas síndromes.
terça-feira, 24 de julho de 2012
Organismos Geneticamente Modificados (ONG´s)
Os organismos geneticamente modificados (OGMs), ou transgênicos, são aqueles que tiveram genes estranhos, de qualquer outro ser vivo, inseridos em seu código genético. O processo consiste na transferência de um ou mais genes responsáveis por determinada característica num organismo para outro organismo ao qual se pretende incorporar esta característica.
Pode-se, com essa tecnologia, inserir genes de porcos em seres humanos, de vírus ou bactérias em milho e assim por diante.
Quase todos os países da Europa têm rejeitado os produtos transgênicos. Devido à pressão de grupos ambientalistas e da população, os governos europeus proibiram sua comercialização e seu cultivo (quase 80% dos europeus não querem consumir transgênicos).
As sementes transgênicas são patenteadas pelas empresas que as desenvolveram. Quando o agricultor compra essas sementes, ele assina um contrato que o proíbe de replantá-las no ano seguinte (prática de guardar sementes, tradicional da agricultura), comercializá-las, trocá-las ou passá-las adiante.
Os EUA, o Brasil e a Argentina concentram 80% da produção mundial de soja, na sua maioria exportada para a Europa e para o Japão. Estes mercados consumidores têm visto no Brasil a única opção para a compra de grãos não transgênicos.
São enormes as pressões que vêm sendo feitas sobre o governo brasileiro pelo lobby das indústrias e dos governos americano e argentino e sobre os agricultores brasileiros, através de intensa propaganda da indústria, para que os transgênicos sejam liberados e cultivados.
Ainda não existem normas apropriadas para avaliar os efeitos dos transgênicos na saúde do consumidor e no meio ambiente e há sérios indícios de que eles sejam prejudiciais. Os próprios médicos e cientistas ainda têm muitas dúvidas e divergências quanto aos riscos dessas espécies. Não existe um só estudo, no mundo inteiro, que prove que eles sejam seguros.
Os produtos contendo transgênicos que estão nas prateleiras de alguns supermercados não são rotulados para que o consumidor possa exercer o seu direito de escolha.
Queremos que antes que se tome uma decisão sobre o cultivo, a comercialização e o consumo de transgênicos no Brasil, sejam feitas pesquisas por instituições científicas de comprovada competência e independência, que assegurem que os transgênicos não são prejudiciais à saúde e ao meio ambiente.
Ao mesmo tempo, queremos que sejam realizadas pesquisas e que haja incentivos para desenvolver a agroecologia - uma agricultura que respeite o meio ambiente e leve em consideração as condições sociais do setor.
Sua ação é orientada por uma coordenação nacional, formada pelas seguintes instituições: ACTION AID - AS-PTA - ESPLAR - GREENPEACE - IDEC - INESC - FASE Nacional.
Queremos que antes que se tome uma decisão sobre o cultivo, a comercialização e o consumo de transgênicos no Brasil, sejam feitas pesquisas por instituições científicas de comprovada competência e independência, que assegurem que os transgênicos não são prejudiciais à saúde e ao meio ambiente.
Ao mesmo tempo, queremos que sejam realizadas pesquisas e que haja incentivos para desenvolver a agroecologia - uma agricultura que respeite o meio ambiente e leve em consideração as condições sociais do setor.
Sua ação é orientada por uma coordenação nacional, formada pelas seguintes instituições: ACTION AID - AS-PTA - ESPLAR - GREENPEACE - IDEC - INESC - FASE Nacional.
Sua ação é orientada por uma coordenação nacional, formada pelas seguintes instituições: ACTION AID - AS-PTA - ESPLAR - GREENPEACE - IDEC - INESC - FASE Nacional.
Sua ação é orientada por uma coordenação nacional, formada pelas seguintes instituições: ACTION AID - AS-PTA - ESPLAR - GREENPEACE - IDEC - INESC - FASE Nacional.
Ao mesmo tempo, queremos que sejam realizadas pesquisas e que haja incentivos para desenvolver a agroecologia - uma agricultura que respeite o meio ambiente e leve em consideração as condições sociais do setor.
Sua ação é orientada por uma coordenação nacional, formada pelas seguintes instituições: ACTION AID - AS-PTA - ESPLAR - GREENPEACE - IDEC - INESC - FASE Nacional.
Sua ação é orientada por uma coordenação nacional, formada pelas seguintes instituições: ACTION AID - AS-PTA - ESPLAR - GREENPEACE - IDEC - INESC - FASE Nacional.
A Campanha "Por um Brasil Livre de Transgênicos"
Os transgênicos ainda estão proibidos no Brasil e o tema ganha dimensão nacional e interesse popular graças às ações das ONGs.
A Campanha Por Um Brasil Livre de Transgênicos foi criada por um grupo de organizações não governamentais (ONGs) preocupadas com as conseqüências que o uso dos transgênicos pode trazer para nossa saúde, para o meio-ambiente e para a economia do País.
DNA Recombinante
Todos os seres vivos apresentam, no interior de suas células, moléculas de DNA e/ou de RNA, chamadas de ácidos nucléicos. Os ácidos nucléicos são as biomoléculas celulares mais importantes, pois contêm o material genético do organismo, composto por uma seqüência de genes (genoma). Os ácidos nucléicos são responsáveis pelo armazenamento e pela transmissão do material genético e pela sua tradução, que resulta na síntese das proteínas.
As proteínas fazem parte da estrutura de um ser vivo e estão relacionadas ao funcionamento do organismo deste ser, ajudando na produção de enzimas e hormônios. Os milhares de organismos existentes na natureza são determinados pelas diferenças protéicas, pois são as proteínas que determinam as características de um organismo, ou seja, o seu fenótipo.
Quando o genoma de uma planta, de um animal ou de um microorganismo é modificado, alterando-se os genes existentes ou incorporando-se genes de outro organismo, as características ou fenótipo desse organismo também se alteram, resultando em um Organismo Transgênico ou Organismo Geneticamente Modificado (OGM). Se estes genes forem herdáveis, a descendência também será alterada.
O organismo GM ou transgênico é “qualquer organismo, com exceção do ser humano, cujo material genético tenha sido modificado de uma forma que não ocorre naturalmente por meio de cruzamentos e/ou recombinação natural” (Artigo 2° da Diretiva 2001/18/CE do Parlamento Europeu e do Conselho da União Européia de 12 de março de 2001).
O avanço da engenharia genética, utilizando técnicas de DNA recombinante, permitiu que genes responsáveis por uma determinada característica favorável fossem identificados, modificados e incorporados a outro organismo de espécie igual ou diferente. Isso resultou em um enorme avanço nos processos naturais de melhoramento de plantas, animais e microorganismos.
Com a tecnologia do DNA recombinante pode-se, de forma rápida, modificar a função de um gene pré-existente numa planta ou incorporar nesta um único gene diferente, correspondente a uma determinada característica que se deseja melhorar. A transferência de genes pode ocorrer entre diferentes espécies, de forma que, por exemplo, uma qualidade presente numa leguminosa pode ser transferida para um cereal, transferência impraticável sem essa tecnologia.
Para os seus defensores, a tecnologia do DNA recombinante é, portanto, uma tecnologia moderna com grande potencial para aumentar a produtividade agrícola, reduzir o impacto ambiental da agricultura, minimizando o uso de pesticidas e melhorar a qualidade nutricional e tecnológica dos alimentos. E os alimentos que ela produz não são, necessariamente, menos seguros para a saúde. Assim como toda tecnologia, a transgenia deve ser avaliada e acompanhada por grupos científicos, órgãos governamentais e de segurança alimentar e organizações de defesa do consumidor.
Enzimas de Restrição
As enzimas de restrição ou também denominadas de endonucleases de restrição, são as ferramentas básicas da engenharia genética, desempenhando função de clivagem (corte) da molécula de DNA em pontos específicos, em reconhecimento a determinadas seqüências de nucleotídeos.
Foram inicialmente descobertas em células bacterianas, relacionadas ao mecanismo de defesa contra DNA exógeno, a exemplo parasitose viral, inativando o potencial infeccioso do organismo invasor a partir da fragmentação do material genético do mesmo.
Hoje em dia, a biologia molecular utiliza centenas de endonucleases capazes de seccionar o duplo filamento polinucleotídico da molécula de DNA em lugares determinados. Dessa forma, submetidos à técnica de eletroforese, os pedaços secionados pelas enzimas passam por análise comparativa de bandas transversais reveladas em um filme, permitindo, por exemplo, a identificação de pessoas: possíveis suspeitos de um crime ou na determinação da paternidade, tendo o laudo legitimidade relativa de competência jurisdicional.
Segue abaixo um quadro demonstrativo de algumas enzimas restritivas com indicação da seqüência de reconhecimento de bases nitrogenadas e o ponto de corte.
Nos exames de paternidade, aplicando-se a eletroforese em fragmentos de DNA, obtidos por ação de enzimas de restrição em amostras sangüíneas dos entes envolvidos: mãe(M), filhos (F1, F2, F3 e F4) e o suposto pai (SP), indicam por bandas eletrondensas que a herança de seqüenciamentos presentes nos filhos e ausentes na mãe, somente pode ter sido transferidos aos filhos pelo pai. No caso exemplificado a seguir, constata-se a contribuição genética provável do suposto pai, indicada por círculos vermelhos no filme revelado da eletroforese.
.jpg)
Engenharia Genética
A Engenharia Genética: conjunto de técnicas que envolvem a manipulação de genes de um determinado organismo, geralmente de forma artificial. Esta manipulação envolve duplicação, transferência e isolamento de genes, com o objetivo de produzir organismos geneticamente melhorados para desempenharem melhor suas funções e produzir substâncias úteis ao homem.

Através da engenharia genética muitos hormônios passaram a ser produzidos por bactérias com DNA modificado, como por exemplo, a insulina, que era produzida por animais e causava alguns efeitos colaterais indesejáveis em seres humanos. O hormônio de crescimento era extraído da hipófise de cadáveres e houve casos de pessoas que se contaminaram com uma doença neurológica chamada Creutzfeldt-Jacob.

Endogamia
Endogamia é um sistema em que os acasalamentos se dão entre indivíduos aparentados, relacionados pela ascendência, ou seja, é a união de indivíduos mais aparentados do que a média da população.

Tem como efeito genético a diminuição da heterozigose e o aumento da homozigose, e, como efeito fenotípico, uma grande manifestação de genes recessivos, que acabam resultando em perda de vigor, assim como a perda da variância, à medida que aumenta o parentesco.

Endogamia pode estar estabelecida em grupos sociais, áreas ou relações. Vários tipos de endogamia foram observados em populações humanas, tal como:
Endogamia de aldeia (por exemplo, nos Ianomâmis)
Endogamia de linhagem (por exemplo, em comunidades pastorais do Médio Oriente)
Endogamia de castas (por exemplo, na Índia)
Endogamia de classes (por exemplo, nos Estados Unidos)
O que é melhoramento genético?
O melhoramento genético é uma ciência utilizada em plantas e animais que visa aumentar a frequência de alelos favoráveis em uma população animal ou vegetal.
Para que seja iniciado um programa de melhoramento é necessário haver variabilidade genética na população, e o progresso do programa será maior tanto quanto for maior essa variabilidade.
Processos de melhoramento


No melhoramento clássico existem duas formas de se melhorar indivíduos: Seleção e Cruzamento.A seleção consiste em escolher os pais da próxima geração que apresentam um maior número de alelos favoráveis (ex.Crescimento,resistência a doenças, produção de sementes, produção de leite, etc.)
O cruzamento consiste em acasalar indivíduos diferentes geneticamente (ex. Bovino Gir x Bovino Holandês) para desfrutar da heterose e complementariedade. Heterose é o fenômeno que ocorre quando o desempenho médios dos filhos é superior a média dos pais, esse fenômeno ocorre porquê a maioria das características desejáveis apresentam caráter dominante, e, ao se cruzar indivíduos puros diferentes, há um aumento no número dos loci em heterozigose (ex. AaBbCcDd) esse fato leva a progênie possuir mais alelos favoráveis.

O "contrário" da heterose seria a endogamia, ou seja indíduos, com grande número de loci em homozigose (ex. AAbbCCddeeffGG). Alguns alelos, principalmente os recessivos, quando em homozigose podem levar o indivíduo a expressar características deletérias, como síndromes, deformidades e diminuição do desempenho das características de baixa herdabilidade, que são controladas por muitos genes, como as características relacionadas a reprodução, resistência a doenças etc.
Os seres vivos possuem vários pares de cromossomos. Cada um destes possui genes que são os responsáveis pela expressão das características dos indivíduos. Na divisão celular que antecede a formação dos gametas masculinos (espermatozóides) e feminino (óvulo) os pares de cromossomos se separam e assim cada um reúne a metade do número de cromossomos da célula original.
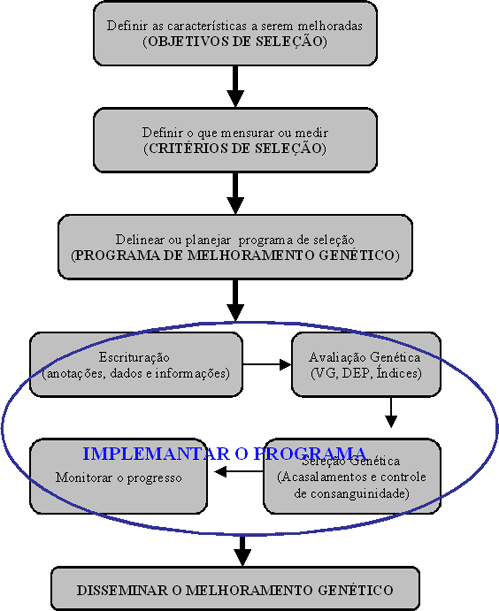
Cada novo indivíduo recebe ao se formar um conjunto de cromossomos do pai e outro da mãe sendo restabelecido o número de cromossomos da espécie. Na reconstituição dos cromossomos, a predominância ou não dos genes para uma mesma característica determinará se ela será expressada ou não. A observação de muitas gerações nos permite ainda identificar características de alta herdabilidade, ou de baixa herdabilidade.
Na medida em que se promove o melhoramento genético por um programa de seleção eficaz, e que este se perpetua por muitas gerações mantendo-se o mesmo critério de seleção, há redução da variabilidade genética como resultado do incremento de homozigose.
Para que seja iniciado um programa de melhoramento é necessário haver variabilidade genética na população, e o progresso do programa será maior tanto quanto for maior essa variabilidade.
Processos de melhoramento
No melhoramento clássico existem duas formas de se melhorar indivíduos: Seleção e Cruzamento.A seleção consiste em escolher os pais da próxima geração que apresentam um maior número de alelos favoráveis (ex.Crescimento,resistência a doenças, produção de sementes, produção de leite, etc.)
O cruzamento consiste em acasalar indivíduos diferentes geneticamente (ex. Bovino Gir x Bovino Holandês) para desfrutar da heterose e complementariedade. Heterose é o fenômeno que ocorre quando o desempenho médios dos filhos é superior a média dos pais, esse fenômeno ocorre porquê a maioria das características desejáveis apresentam caráter dominante, e, ao se cruzar indivíduos puros diferentes, há um aumento no número dos loci em heterozigose (ex. AaBbCcDd) esse fato leva a progênie possuir mais alelos favoráveis.
O "contrário" da heterose seria a endogamia, ou seja indíduos, com grande número de loci em homozigose (ex. AAbbCCddeeffGG). Alguns alelos, principalmente os recessivos, quando em homozigose podem levar o indivíduo a expressar características deletérias, como síndromes, deformidades e diminuição do desempenho das características de baixa herdabilidade, que são controladas por muitos genes, como as características relacionadas a reprodução, resistência a doenças etc.
Os seres vivos possuem vários pares de cromossomos. Cada um destes possui genes que são os responsáveis pela expressão das características dos indivíduos. Na divisão celular que antecede a formação dos gametas masculinos (espermatozóides) e feminino (óvulo) os pares de cromossomos se separam e assim cada um reúne a metade do número de cromossomos da célula original.
Cada novo indivíduo recebe ao se formar um conjunto de cromossomos do pai e outro da mãe sendo restabelecido o número de cromossomos da espécie. Na reconstituição dos cromossomos, a predominância ou não dos genes para uma mesma característica determinará se ela será expressada ou não. A observação de muitas gerações nos permite ainda identificar características de alta herdabilidade, ou de baixa herdabilidade.
Na medida em que se promove o melhoramento genético por um programa de seleção eficaz, e que este se perpetua por muitas gerações mantendo-se o mesmo critério de seleção, há redução da variabilidade genética como resultado do incremento de homozigose.
Biotecnologia
Biotecnologia é tecnologia baseada na biologia, especialmente quando usada na agricultura, ciência dos alimentos e medicina. A Convenção sobre Diversidade Biológica da ONU possui uma das muitas definições de biotecnologia:
"Biotecnologia define-se pelo uso de conhecimentos sobre os processos biológicos e sobre as propriedades dos seres vivos, com o fim de resolver problemas e criar produtos de utilidade."

A definição ampla de biotecnologia é o uso de organismos vivos ou parte deles, para a produção de bens e serviços. Nesta definição se enquadram um conjunto de atividades que o homem vem desenvolvendo há milhares de anos, como a produção de alimentos fermentados (pão, vinho, iogurte, cerveja, e outros). Por outro lado a biotecnologia moderna se considera aquela que faz uso da informação genética, incorporando técnicas de DNA recombinante.
A biotecnologia combina disciplinas tais como genética, biologia molecular, bioquímica, embriologia e biologia celular, com a engenharia química, tecnologia da informação, robótica, bioética e o biodireito, entre outras.
Medusa Van Allen x Pequena Miss Sunshine
Quando nasceu em 19 de março de 1908, Medusa Van Allen, parecia uma criança normal. Mas com o passar do tempo sua mãe começou a achar estranho o fato de seu corpo sempre permanecer como o do dia que tinha nascido. A partir daí ela passou por um calvário à busca de médicos para saber o que a filha tinha. Nunca descobriram qual era asua doença.

Com a aparência bizarra acabou sendo mais uma atração, "Pequena Miss Sunshine", dos circos Ripley de Ohio, estado onde nasceu e morava. Ela cantava, dublava e recitava poemas.
No final da década de 40, Medusa –este era realmente seu verdadeiro nome– já havia conseguido economizar uma razoável quantia em dinheiro, que utilizou para a contratação de professores particulares, quando aprendeu Economia e resolveu investir nesta área tendo um resultado satisfatório como empreendedora.

Bem por essa época o interesse público sobre ela era bem maior que quando trabalhava no circo. Meio a contra-gosto resolveu escrever uma carta para ser publicada no jornal de maior tiragem da época. O texto da carta era o seguinte:
"Queridos leitores.
Com considerável relutância eu relatarei a breve história de minha vida. Eu repudio a idéia de falar de assuntos íntimos e pessoais, mas as minhas circunstâncias o fazem necessário, mas é deprimente que o interesse do público vise mais a minha análise física do que pessoal.
Eu nasci no dia 19 de março de 1908, fui criada no Estado de Ohio. Nasci com uma condição que fez com que os ossos de meu corpo não amadurecessem, apenas minha cabeça "envelheceria".
Estou com 40 anos e meu corpo ainda é o mesmo de quando eu era um bebe. Passei boa parte de meus dias em clínicas, porém nunca houve esperança de cura para mim e todos diziam que eu viveria pouco tempo.
Eu não posso unir minhas mãos, não posso tocar em meu rosto, mas isso não me desestimulou em tentar viver uma vida normal. Adquiri uma boa educação, tive tutores particulares que me ensinaram e me estimularam, consigo escrever cartas aos amigos, também consegui êxito estudando economia e hoje dirijo meus próprios negócios.
Eu desfruto da vida como qualquer pessoa normal, faço tudo com prazer e acredito que um sorriso e palavras cordiais resolvem qualquer problema.
Sinto-me demasiadamente contente por estar com boa saúde e contando com muitos amigos. O mundo é bom, cheio de pessoas boas, a maioria delas está pronta para fazer boas ações. Então, porque irei deixar de sorrir?
Medusa Van Allen"

Não há registros da data de seu falecimento e ficaram mais perguntas que respostas desta mulher surpreendente. Que doença afinal tinha? Seu relato supôe que teriaOsteogénese imperfeita, mas as pessoas que sofrem deste mal geralmente podem sentar e usar cadeira de rodas. Além disso, a foto onde ela aparece no colo da mãe induz que Meduza tinha o corpo todo teso e não mole como diz na carta.
Frank Lentini
Ele nasceu em 1889 em Siracusa (Sicília) em uma família com onze filhos. Foi levado por sua tia, ainda bebê, para uma orfanato de inválidos, depois que seus pais recusaram-se a reconhecê-lo como filho.

Lentini nasceu com três pernas, dois órgãos genitais e um pé no joelho da terceira perna. Assim, no total, ele tinha três pernas, quatro pés, dezesseis dedos dos pés, e dois órgãos genitais funcionais que eram tudo o que restou de um gêmeo parasita.

Os médicos decidiram pela não remoção dos órgãos pois poderia resultar em paralisia e até a morte. Quando tinha nove anos de idade Frank deixou o orfanato de crianças inválidas na qual viveu certo período e foi levado para os EUA para ser exibido em circos de aberrações.

Trabalhou no Ringling Brothers Circus fazendo um espetáculo de grande sucesso chamado "O Jogador de Futebol de Três Pernas". O que mais atraia o público era a maneira desengonçada de Frank ao se locomover com a bola, devido à diferença de comprimento entre suas três pernas.

Apesar das adversidades, Frank nunca foi uma pessoa ressentida com sua deformidade, ele se mostrava orgulhoso por sua terceira perna e via nessa condição uma vantagem e não uma infelicidade. Estranhamente a única coisa que o incomodava em seu corpo era um dedo polegar extra que apareceu em um joelho de uma de suas pernas, Frank sempre procurou esconder esse dedão.

Em 1930 Frank Lentini se tornou oficialmente um cidadão americano, nessa mesma década conheceu Thereza Murray, paixão fulminante que terminou em casmento que gerou quatro filhos.

Foi reconhecido como um exímio jogador e conseguiu ganhar algum dinheiro com suas apresentações para os atletas das ligas de futebol. Seus filhos o descreveram como um pai presente, atencioso e muito amoroso, mas que no fundo sempre carregou certa tristeza por ter sido um dia rejeitado por seus próprios pais.

Lentini morreu na sua casa em Jacksonville, Flórida no dia 22 de setembro de 1966.

Fenilcetonuria
O que é Fenilcetonúria?
A fenilcetonúria (PKU) é uma doença rara na qual o bebê nasce sem a habilidade de quebrar adequadamente um aminoácido chamado fenilalanina.

Causas:
A fenilcetonúria é hereditária, isto é, passa de pais para filhos. O pai e a mãe devem passar o gene defeituoso para que o bebê tenha essa doença. Isso é conhecido como traço recessivo autossômico.
Os bebês com PKU não possuem uma enzima chamada fenilalanina hidroxilase, necessária para quebrar um aminoácido essencial denominado fenilalanina. Essa substância é encontrada em alimentos que contêm proteínas.
Sem essa enzima, os níveis de fenilalanina e de duas substâncias associadas a ela crescem no organismo. Tais substâncias são prejudiciais ao sistema nervoso central e causam dano cerebral.

Exames:
A fenilcetonúria pode ser fácilmente detectada em um simples exame de sangue. Em todos os estados brasileiros, o exame de triagem para PKU (ou fenilcetonúria), chamado teste do pezinho, é exigido para todos os recém-nascidos como parte do painel de triagem. O teste normalmente é realizado por meio da retirada de algumas gotas de sangue do bebê antes da saída dele do hospital.
Se o teste inicial é positivo, exames de sangue e urina são feitos para confirmar o diagnóstico.
Galactosemia
A galactosemia pode ser definida como uma exacerbada concentração sanguínea do monossacarídeo galactose (aldohexose, epímera da glicose em C-4), resultante de uma desordem no metabolismo gerado por deficiente atividade enzimática ou função hepática alterada.

A espécie humana adquire a galactose inicialmente por meio do leite humano e bovino e de derivados lácteos, por meio da quebra (hidrólise) da lactose (glicose+galactose ligadas por uma ponta β-glicosídica). A galactose livre também é encontrada em vegetais, como banana, maçã, tomate, entre outros. A digestão da lactose é intermediada pela lactase (enzima intestinal), responsável por hidrolisar os monossacarídeos que a formam. Subseqüentemente à quebra da lactose em glicose e galactose, ocorre o processo de metabolização desses monossacarídeos, englobando catálise enzimática que irá levar, por fim, à conversão da galactose em glicose (fonte de energia). O processo de metabolização da galactose encontra-se alterada em pacientes galactosêmicos, em conseqüência das deficiências enzimáticas em diversos níveis.
Esta doença apresenta origem genética, condicionada por um gene autossômico recessivo. Pesquisas realizadas desde 1979 apontam que 1 entre 7.500 nascidos vivos apresentará alguma forma de galactosemia. Acredita-se também que 1 em cada 40 indivíduos carrega o gene defeituoso. A galactosemia clássica acomete 1 entre cada 60.000 nascidos vivos.
O defeito no metabolismo da galactose na doença em questão é causado principalmente pela deficiência em três enzimas que participam da via metabólica da galactoses, são elas: galactoquinase, galactose-1-P-uridil transferase e uridina-difosfogalactose epimerase.
Em casos de deficiência enzimática, a galactose segue uma via metabólica alterada, levando a seu acúmulo, o que acarreta uma reação de conversão de galactose em galactitol, um poliálcool altamente tóxico, ou em galactonato, que também é tóxico.
As manifestações clínicas apresentadas pelos pacientes galactosêmicos são vômito, hepatomegalia (aumento do fígado), falhas renais, danos cerebrais, falhas ovarianas, deficiência de aprendizagem e galactosuria.
O aumento hepático ocorre devido ao acúmulo de galactitol nos hepatócitos, resultando em aumento da pressão osmótica e conseqüente entrada de água nas células, deixando-as hipertróficas.
Os problemas oculares resultam de um acúmulo de produtos tóxicos que levam à formação da catarata, podendo evoluir para perda de visão total.
Os problemas neurológicos não estão correlacionados apenas com acúmulo de substâncias tóxicas, apresentam causas mais complexas.

Existem diferentes tipos de galactosemia, dentre eles estão:
Galactosemia do Tipo 1: é a forma mais comum e mais grave. É causada pela deficiência da galactose-1-P uridil transferase (GALT). A ausência completa dessa enzima é denominada galactosemia clássica, mais comumente conhecida apenas pelo termo “galactosemia”. Indivíduos portadores dessa forma que não são tratados devidamente podem apresentar problemas renais e hepáticos, catarata, problemas neurológicos e insuficiência ovariana precoce.
Variante Duarte: atividade parcial da GALT ocorre em diferentes variantes, sendo a Duarte a mais comum, na qual o indivíduo apresenta um alelo Duarte e um alelo clássico, levando a uma atividade enzimática correspondente a 25% da normal. Pacientes com dois alelos Duarte possuem cerca de 50% de atividade enzimática normal.
Galactosemia do Tipo 2: neste tipo há um defeito da enzima galactoquinase, o que resulta no acúmulo de galactose, apresentando como características marcantes os problemas oculares.
Galactosemia do Tipo 3: este tipo é causada pelo defeito da enzima uridil difosfo galactose-4-epimerase. Esta forma é rara.
Outros tipos: existem tipos de galactosemia que são benignos e assintomáticos.
Normalmente, o diagnóstico da galactosemia é feito por meio de um teste enzimático, demonstrando que a atividade da galactose-1-P uridil transferase está ausente ou em níveis muito reduzidos nos eritrócitos (no caso da galactose clássica). O diagnóstico pré-natal pode ser realizado por meio da amniocentese, com cultura de fibroblastos oriundos do líquido amniótico. O teste de tolerância à galactose é contra-indicado, pois pode ser fatal em pacientes com galactosemia.
O tratamento é feito por meio de um controle alimentar, evitando a ingestão de galactose e lactose na dieta. Como existem diversas formas de galactosemia, as restrições na dieta irão variar de acordo com cada tipo. Nos casos mais severos deve ser realizado um monitoramente multidisciplicar, objetivando minimizar as conseqüências da enfermidade.
Hipotiroidismo Congênito
O hipotireoidismo congênito (HC) é um mal hereditário que impossibilita o organismo de gerar o hormônio tireoidiano T4, impedindo o crescimento e desenvolvimento do recém-nascido. O HC é a causa mais comum de retardo mental.
É possível diagnosticar a doença e iniciar o tratamento evitando suas conseqüências, porém é preciso que seja feito na primeira semana de nascimento, por isso, a realização do Teste do Pezinho é fundamental. A triagem neonatal detecta o HC. No Brasil, o Teste do Pezinho é obrigatório e em estados, como Santa Catarina, a abrangência é de 100% dos nascidos.

A cura inclui administração de hormônio tireoidiano, sob rigoroso controle médico, para que o bebê fique bom e tenha uma vida normal. Cerca de um a cada 4.000 recém-nascidos possuem esse distúrbio.
O HC ocorre, mundialmente, em 1/3000-4000 neonatos e pode ser classificado em permanente ou transitório. O HC primário é responsável pela maioria dos afetados, enquanto o secundário e terciário são raros. Nos países iodo-suficientes, a disgenesia tireóidea (DT) é a causa mais freqüente de HC.

Os defeitos hereditários da síntese hormonal ocorrem em minoria de crianças portadoras de HC. Fatores ambientais, genéticos e auto-imunes concorrem na etiologia do HC, mas na maioria dos casos de DT a causa é obscura. Atribui-se aos genes envolvidos na ontogenia da glândula tireóidea, como os fatores de transcrição TITF1, TITF2, PAX-8 e receptor de TSH (TSHR), função patogenética na DT. Até o momento não foi descrita anormalidade no gene TITF1 como causa de HC, enquanto foram identificadas mutações no PAX-8 em cinco recém-nascidos com DT. Embora não envolvidas na DT, mutações inativadoras do TSHR podem produzir espectro de defeitos congênitos oscilando entre hipertirotropinemia com eutireoidismo e hipotireoidismo com hipoplasia glandular. A clonagem dos genes envolvidos na biossíntese dos hormônios tireóideos, como o da tireoperoxidase (TPO) e tireoglobulina (Tg), permitiu a identificação de mutações responsáveis por alguns casos de bócio e hipotireoidismo decorrente de defeito de incorporação de iodeto ou anormalidades na síntese de Tg. Recentemente, foi demonstrada a base molecular do defeito de transporte ativo de iodeto e da síndrome de Pendred, respectivamente, devidas a mutações no gene NIS (simportador de sódio e iodeto) e no gene PDS (pendrina). Em conclusão, grande parte dos pacientes com HC e DT não tem esclarecida, ainda, a causa molecular desta síndrome.
Teste do Pezinho
O Teste do Pezinho é um exame laboratorial simples que tem o objetivo de detectar precocemente doenças metabólicas, genéticas e ou infeciosas que poderão causar lesões irreverssíveis no bebê, como por exemplo retardo mental. A maioria das doenças pesquisadas podem ser tratadas com sucesso desde que diagnósticadas antes mesmo de manifestar os primeiros sintomas. Existem três tipos de teste: Básico, Ampliado e Plus.

MÉTODO:
O exame ficou popularmente conhecido como “teste do pezinho” por ser realizado através da análise de amostras de sangue coletadas através do calcanhar do bebe. É um procedimento simples que não traz riscos para a criança.

RESULTADOS:
O resultado será enviado no prazo Médio de 7 a 10 dias úteis para a mãe e o pediatra.

QUANDO FAZER:
O período ideal para a realização da coleta do Teste do Pezinho é a partir do 3º dia de vida do bebê ou o mais brevemente possível. Isto não invalida, entretanto, a sua realização em bebês com mais dias de vida. O que poderá ser prejudicada é a eficácia do tratamento, caso necessário.
O teste do pezinho, ou triagem neonatal como também é chamado, é um exame obrigatoriamente realizado em todos os bebês recém-nascidos, entre o 3º e o 7º dia de vida. As doenças detectadas pelo teste do pezinho ampliado geralmente são:

MÉTODO:
O exame ficou popularmente conhecido como “teste do pezinho” por ser realizado através da análise de amostras de sangue coletadas através do calcanhar do bebe. É um procedimento simples que não traz riscos para a criança.
RESULTADOS:
O resultado será enviado no prazo Médio de 7 a 10 dias úteis para a mãe e o pediatra.
QUANDO FAZER:
O período ideal para a realização da coleta do Teste do Pezinho é a partir do 3º dia de vida do bebê ou o mais brevemente possível. Isto não invalida, entretanto, a sua realização em bebês com mais dias de vida. O que poderá ser prejudicada é a eficácia do tratamento, caso necessário.
O teste do pezinho, ou triagem neonatal como também é chamado, é um exame obrigatoriamente realizado em todos os bebês recém-nascidos, entre o 3º e o 7º dia de vida. As doenças detectadas pelo teste do pezinho ampliado geralmente são:
Talassemia
A talassemia (palavra oriunda do grego thalassa=mar; haemas=sangue), também conhecida como anemia de Cooley(primeiro médico que descreveu a doença em 1925), é uma doença hereditária autossômica recessiva que acomete o sangue.

Caracteriza-se por uma anemia leve ou severa, decorrente da hemoglobina reduzida e menor quantidade de eritrócitos(células vermelhas) do que o normal. Em indivíduos portadores da talassemia, os genes que codificam a hemoglobina (proteína dos eritrócitos que carregam o oxigênio) estão faltando ou são diferentes.
Existem dois tipos de talassemia:
α-talassemia: quando o defeito genético está na cadeia α da hemoglobina.
β-talassemia: quando o defeito genético está na cadeia β da hemoglobina.

Esse defeito genético tem maior prevalência em populações do sudoeste da Ásia e China (α-talassemia), e também, Grécia e Itália (β-talassemia). Por predominar em populações que vivem em torno do Mediterrâneo recebem também o nome de anemia do Mediterrâneo.
Por ser uma doença hereditária, é transmitida de geração para geração. O quadro clínico apresentado por indivíduos portadores desses genes é muito variável, dependendo da carga genética, se homozigótica ou heterozigótica. Simplificadamente pode-se dividir em dois quadros clínicos completamente distintos:
Talassemias maiores: esse quadro é mais raro, a anemia é severa, iniciando-se nos primeiros meses de vida do indivíduo, acompanhada de icterícia, deformidades ósseas e esplenomegalia.
Talassemias menores: há a presença de discreta anemia, com a qual o indivíduo pode conviver normalmente, sendo que algumas vezes nem anemia existe.
Na talassemia menor, a suspeita é levantada quando se realiza a investigação de uma discreta anemia em um indivíduo. No caso da talassemia maior, o diagnóstico é feito já nos primeiros meses de vida, em consequência da grave anemia. Em ambas as formas, exames laboratoriais que estudam a hemoglobina sanguínea, como a cromatografia da hemoglobina, em associação com as formas da hemácia fazem o diagnóstico.
Na maioria das vezes, a talassemia menor não necessita de tratamento. Entretanto, em certas situações, como gravidez, recomenda-se a suplementação de ácido fólico na dieta. É importante evitar a ingestão de sais de ferro, uma vez que a talassemia está associada a uma maior absorção de ferro da dieta. Essa ingestão excessiva pode resultar em exacerbado acúmulo de ferro.
O tratamento da talassemia maior é bem mais complicado. Inclui programa de transfusão sanguínea permanente, esplenectomia e uso de quelantes para retirar excesso de ferro resultantes das inúmeras transfusões. Quando o paciente dispõe de um doador de medula compatível, esse tipo de procedimento pode ser indicado.
Caracteriza-se por uma anemia leve ou severa, decorrente da hemoglobina reduzida e menor quantidade de eritrócitos(células vermelhas) do que o normal. Em indivíduos portadores da talassemia, os genes que codificam a hemoglobina (proteína dos eritrócitos que carregam o oxigênio) estão faltando ou são diferentes.
Existem dois tipos de talassemia:
α-talassemia: quando o defeito genético está na cadeia α da hemoglobina.
β-talassemia: quando o defeito genético está na cadeia β da hemoglobina.
Esse defeito genético tem maior prevalência em populações do sudoeste da Ásia e China (α-talassemia), e também, Grécia e Itália (β-talassemia). Por predominar em populações que vivem em torno do Mediterrâneo recebem também o nome de anemia do Mediterrâneo.
Por ser uma doença hereditária, é transmitida de geração para geração. O quadro clínico apresentado por indivíduos portadores desses genes é muito variável, dependendo da carga genética, se homozigótica ou heterozigótica. Simplificadamente pode-se dividir em dois quadros clínicos completamente distintos:
Talassemias maiores: esse quadro é mais raro, a anemia é severa, iniciando-se nos primeiros meses de vida do indivíduo, acompanhada de icterícia, deformidades ósseas e esplenomegalia.
Talassemias menores: há a presença de discreta anemia, com a qual o indivíduo pode conviver normalmente, sendo que algumas vezes nem anemia existe.
Na talassemia menor, a suspeita é levantada quando se realiza a investigação de uma discreta anemia em um indivíduo. No caso da talassemia maior, o diagnóstico é feito já nos primeiros meses de vida, em consequência da grave anemia. Em ambas as formas, exames laboratoriais que estudam a hemoglobina sanguínea, como a cromatografia da hemoglobina, em associação com as formas da hemácia fazem o diagnóstico.
Na maioria das vezes, a talassemia menor não necessita de tratamento. Entretanto, em certas situações, como gravidez, recomenda-se a suplementação de ácido fólico na dieta. É importante evitar a ingestão de sais de ferro, uma vez que a talassemia está associada a uma maior absorção de ferro da dieta. Essa ingestão excessiva pode resultar em exacerbado acúmulo de ferro.
O tratamento da talassemia maior é bem mais complicado. Inclui programa de transfusão sanguínea permanente, esplenectomia e uso de quelantes para retirar excesso de ferro resultantes das inúmeras transfusões. Quando o paciente dispõe de um doador de medula compatível, esse tipo de procedimento pode ser indicado.
Hemofilia
Hemofilia é uma doença genético-hereditária que se caracteriza por desordem no mecanismo de coagulação do sangue e manifesta-se quase exclusivamente no sexo masculino.
 Existem dois tipos de hemofilia: A e B. A hemofilia A ocorre por deficiência do fator VIII de coagulação do sangue e a hemofilia B, por deficiência do fator IX.
Existem dois tipos de hemofilia: A e B. A hemofilia A ocorre por deficiência do fator VIII de coagulação do sangue e a hemofilia B, por deficiência do fator IX.
A doença pode ser classificada, ainda, segundo a quantidade do fator deficitário em três categorias: grave (fator menor do que 1%), moderada (de 1% a 5%) e leve, acima de 5%. Neste caso, às vezes, a enfermidade passa despercebida até a idade adulta.
Causa:
O gene que causa a hemofilia é transmitido pelo par de cromossomos sexuais XX. Em geral, as mulheres não desenvolvem a doença, mas são portadoras do defeito. O filho do sexo masculino é que pode manifestar a enfermidade.
Doença de Alzheimer
A doença de Alzheimer (Alois Alzheimer, neurologista alemão que primeiro descreveu essa patologia) provoca progressiva e inexorável deterioração das funções cerebrais, como perda de memória, da linguagem, da razão e da habilidade de cuidar de si próprio.

Cerca de 10% das pessoas com mais de 65 anos e 25% com mais de 85 anos podem apresentar algum sintoma dessa enfermidade e são inúmeros os casos que evoluem para demência. Feito o diagnóstico, o tempo médio de sobrevida varia de 8 a 10 anos.
Causas:
Não se conhece a causa específica da doença de Alzheimer. Parece haver certa predisposição genética para seu aparecimento. Nesses casos, ela pode desenvolver-se precocemente, por volta dos 50 anos.
Pesquisadores levantam a hipótese de que algum vírus e a deficiência de certas enzimas e proteínas estejam envolvidos na etiologia da doença. Outros especulam que a exposição ao alumínio e seu depósito no cérebro possam contribuir para a instalação do quadro, mas não foi estabelecida nenhuma relação segura de causa e efeito a respeito disso.
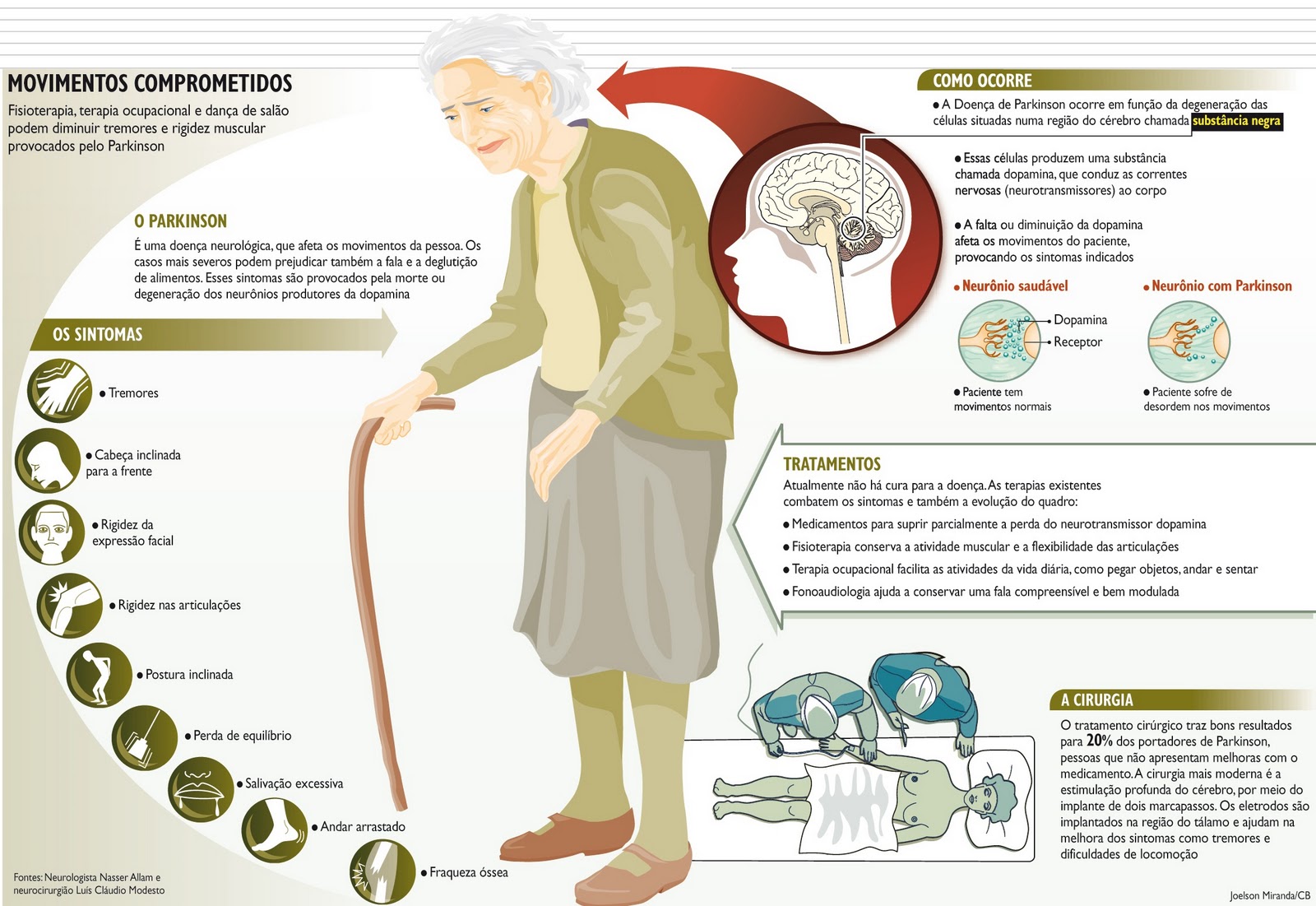
Doença de Parkinson
Doença neurológica, sem causa conhecida, que atinge o sistema nervoso central e compromete os movimentos. Quanto maior a faixa etária, maior a incidência da doença de Parkinson. De acordo com as estatísticas, na grande maioria dos pacientes, ela surge a partir dos 55, 60 anos e sua prevalência aumenta a partir dos 70, 75 anos.

Sintomas:
Os sintomas da doença de Parkinson variam de um paciente para o outro. Em geral, no início, eles se apresentam de maneira lenta, insidiosa, e o paciente tem dificuldade de precisar a época em que apareceram pela primeira vez.
A lentificação dos movimentos e os tremores nas extremidades das mãos, muias vezes notados apenas pelos amigos e familiares, costumam ser os primeiros sinais da doença. A diminuição do tamanho das letras ao escrever é outra característica importante.
Outros sintomas podem estar associados ao início da doença: rigidez muscular; acinesia (redução da quantidade de movimentos), distúrbios da fala, dificuldade para engolir, depressão, dores, tontura e distúrbios do sono, respiratórios, urinários.
Tratamento:
O tratamento pode ser medicamentoso, psicoterápico e até cirúrgico em alguns casos.
O tratamento medicamentoso é feito à base de drogas neuroprotetoras que visam a evitar a diminuição progressiva de dopamina, neurotransmissor responsável pela transmissão de sinais na cadeia de circuitos nervosos.
O tratamento psicoterápico ocorre em função da depressão, perda de memória e do aparecimento de demências e pode incluir a prescrição de medicamentos antidepressivos e de outros psicotrópicos.
Recomendações:
* Procure um médico tão logo perceba um ligeiro tremor nas mãos ou tenha notado que sua letra diminuiu de tamanho (micrografia);
* Mantenha a atividade intelectual;
* Não atribua ao passar dos anos, a perda da expressão facial e o piscar dos olhos menos frequentes;
* Pratique atividade física.
Doença de Tay Sachs

A doença de Tay-sachs é uma enfermidade causada pela disfunção dos lisossomos, organelas responsáveis pela digestão celular. Resulta de um defeito na hexosaminidase A, enzima que catalisa uma das etapas da digestão intracelular de um lipídio abundante nas membranas das células nervosas, o gangliosídio.

PROBLEMAS OFTALMOLÓGICOS GERADOS PELA DOENÇA
Trata-se de uma doença oriunda de uma herança autossômica recessiva, ou seja, o indivíduo só desenvolve a doença quando herdar os genes defeituosos tanto do pai quanto da mãe. Aqueles que recebem os genes recessivos de apenas um dos genitores não desenvolvem a doença, mas são portadores de genes mutados, e caso tenha filhos com outro portador, os filhos terão a doença de Tay-sachs.

FOTO DE MENINA COM OS SINTOMAS DA DOENÇA
Os sintomas da doença começam a se manifestar ainda no primeiro ano de vida do indivíduo. Por ser uma doença neurodegenerativa, a criança tem seu sistema nervoso bastante comprometido, principalmente no que concerne à capacidade psicomotora. Um dos sinais mais característicos da doença de Tay-Sachs é o aparecimento de uma mancha vermelha no olho, seguida de cegueira, surdez, incapacidade de engolir, atrofia dos músculos e paralisia. Em alguns casos, essa mancha pode não aparecer no início do quadro, o que não descarta o diagnóstico da doença, visto que ela pode surgir posteriormente.
Itabaianinha (Cidade dos anões)
A 120 km de Aracaju, Itabaianinha, com 32.00 habitantes, possui cerca de 80 itabaianinhenses adultos com menos de 1,30 metro. Há pelo menos 200 anos, é comum ver uma porrada de anões caminhando pela cidade. E detalhe, o motivo é por causa do incesto inevitável.
A cidade é praticamente toda rural e cercada por montanhas e por estradas de difícil acesso, tornando comuns os casamentos consangüíneos entre as famílias isoladas. A união entre primos com primas, tios com sobrinhas, é a maneira como encaram a questão amorosa. Apesar dos preconceitos contra os anões e além das intermináveis lendas, eles também amam!
Itabaianinha causou tanto interesse que foi tema do documentário Terra de Gigantes, de Ana Paula Teixeira, e hoje é famosa por receber cientistas norte americanos em busca de explicações para o caso. Existe inclusive uma ministração de hormônios aos anões mais novos para que voltem a crescer. Porém, há aqueles que recusam o medicamento e preferem continuar carregando apelidos: Zé Miúdo, Mundinho, Joaninha, Lerinho. Essa é a Itabaianinha, a cidade do inho.
Adrenoleucodistrofia (ALD)
A adrenoleucodistrofia (ALD), é uma enfermidade de origem genética, rara, englobada dentro do grupo das leucodistrofias, responsável por afetar o cromossomo X, sendo esta uma herança ligada ao sexo de caráter recessivo transmitida pelas mulheres portadoras e que acomete quase que exclusivamente os homens.
Esta doença caracteriza-se por uma alteração do metabolismo dos peroxissomos, resultando em um acúmulo de ácidos graxos de cadeia altamente longa (AGCML) formados por 24 a 26 átomos de carbono no organismo, especialmente no cérebro e nas glândulas adrenais (também chamada de supra-renais). Este acúmulo está relacionado ao processo de desmielinização dos axônios acometendo as transmissões dos impulsos nervosos e a insuficiência adrenal.
 Existem diferentes formas da doença, que são:
Existem diferentes formas da doença, que são:Neonatal: este tipo manifesta-se nos primeiros meses de vida. Os genes afetados responsáveis pela forma neonatal da ALD, não se localizam no cromossomo Y, ou seja, afeta tanto indivíduos do sexo masculino quanto do feminino. Neste caso, os portadores apresentam um tempo de sobrevida de 5 anos. O quadro caracteriza-se por retardo, disfunção adrenal, deterioração neurológica, degeneração da retina, convulsões, hipertrofia hepática, anomalias faciais e musculatura fraca.
Clássica ou infantil: esta é a forma mais grave da ALD, sendo apresentada por aproximadamente 35% dos portadores da doença. Manifesta-se entre os 4 a 10 anos de idade, sendo que o tempo de sobrevida gira ao redor dos 10 anos. A sintomatologia apresentada por esses pacientes são: problemas de percepção; disfunção adrenal; perda de memória, da visão, da audição, da fala; problemas nos movimentos de marcha; demência severa.

Adulta: esta forma é mais leva do que a clássica, manifestando-se no início da adolescência ou no início da idade adulta. Este tipo apresenta sobrevida de décadas. O quadro clínica caracteriza-se por dificuldade de deambulação, disfunção adrenal, impotência, incontinência urinária e deterioração neurológica.
ALD em mulheres: embora esta doença manifeste-se especialmente nos homens, mulheres portadoras também podem apresentar uma forma leve de ALD, apresentando sintomas como ataxia, fraqueza ou paralisia das pernas.
Os tratamentos para esta afecção são complicados ou ineficientes para sua cura completa. O mais conhecido é o azeite de Lorenzo, que foi desenvolvido pelo pai de um menino portador da doença, história que foi retratada no filme “Lorenzo’s Oil” (O óleo de Lorenzo). Esta terapia consiste em introduzir na dieta dos pacientes um composto de azeites. Existe também a opção do transplante de medula óssea, sendo este ainda o mais eficaz.
Síndrome do Triplo X (Trissomia do Triplo X)
A trissomia do triplo X é uma anomalia cromossómica numérica, isto é, é uma alteração ao nível do genoma normal de um indivíduo, com a adição de um cromossoma sexual X extra. São resultantes de uma não disjunção no momento da meiose tanto materna como paterna.
A trissomia do X (47, XXX) ou síndrome do triplo X só ocorre em mulheres, sendo elas reconhecidas assim, como super fêmeas. As portadoras dessa doença genética são fenotipicamente normais, não apresentando assim nenhuma diferença ou aberração na sua aparência física. Nas células 47, XXX, dois dos cromossomas X são inactivados e de replicação tardia. A síndrome do triplo X é uma aberração cromossómica numérica que atinge 1 em cerca de 800 a 1.000 mulheres.
Como sugere o nome, a anomalia confere ao portador um ou mais cromossomas X extra. Existem três tipos principais de ocorrência desta anomalia:
47,XXX. é a mais comum (1:1000-2000);
48;XXX, possuem um retardamento mental mais acentuado;
49,XXXXX, possuem as mesmas características dos triplo e tetra X. Porém como são penta X, possuem um retardamento mental mais acentuado, pois quanto maior o número de X maior será o retardamento mental.
Sintomas e modificações:
Algumas pacientes podem ter convulsões epilépticas. Num lar para pacientes epilépticos, 2 de 209 pacientes tinham o cariótipo XXX. Não se pode determinar definitivamente o quanto o cariótipo XXX aumenta a propensão a psicoses, mas alguns autores avaliam a taxa de psicoses tipo esquizofrenia como sendo aumentada três vezes.
Os estudos de acompanhamento mostraram que as mulheres XXX sofrem as alterações da puberdade numa idade apropriada, mas há relatos de puberdade precoce em certas pacientes. Algumas deram à luz crianças, e estas são praticamente todas cromossomicamente normais. Há défice significativo do desempenho em testes de QI, e cerca de 70% dos pacientes têm problemas de aprendizagem graves.
As mulheres portadoras dessa síndrome apresentam um cromossoma X a mais, apresentando um cariótipo com 47 cromossomas: 47 XX X. Quase todos os erros relacionados à essa síndrome ocorrem durante a ovulogénese pela não disjunção dos cromossomas. Quanto mais cromossomas X a mulher possuir, maior será o índice de retardo mental que ela possuirá. Também conhecida como Síndrome de Jacob, a síndrome do triplo X tem como característica não demonstrar sintomas. As mulheres portadoras podem dar origem a crianças perfeitamente normais. As crianças que possuem essa doença não apresentam os sintomas logo após o nascimento, mas podem apresentar baixo peso. Esta síndrome só ocorre em mulheres, nas quais são fenotipicamente normais, não apresentando assim, nenhuma diferença na sua aparência física.

Testes de diagnóstico:
Algumas mulheres com trissomia do X são identificadas em clínicas de infertilidade e outras em instituições para retardados mentais, mas provavelmente muitas permanecem sem diagnóstico.
Curiosidades:
A trissomia do X e as síndromes mais raras de tetrassomias do X (48,XXXX) e pentassomia do X (49,XXXXX) são os equivalentes na mulher da síndrome de Klinefelter masculina.
A síndrome de tetrassomia do X está associada a atraso mais grave do desenvolvimento físico e mental, e a síndrome de pentassomia do X, assim como o XXXXY, geralmente inclui grande retardo do desenvolvimento com múltiplos defeitos físicos que lembram a síndrome de Down.
A primeira mulher conhecida com a trissomia do triplo X chamava-se Patrícia A. Jacobs no hospital de zona oesta de Edinburgh na Escócia, no ano de 1959. Foi encontrada ao seus 35 anos de idade, medindo 1,76 metros e pesando 58kg, esta mulher tinha ovulação prematura aos 19 anos de idade. Na data da sua concepção os seus pais tinham ambos 40 anos de idade, o que pode explicar o aparecimento desta não-dinjunção cromossómica.
Assinar:
Postagens (Atom)